

Research Areas
Our group researches nanomaterials and nanostructures. We study the structural, dynamic, electronic and magnetic properties of ceramics, nanocrystals, nanotubes, clusters and nano-scale magnetic structures. The principal methodology for studying this kind of problem is by means of computational simulation, particularly, computation of the electronic structure based on density functional theory, as well as classical molecular dynamics and ab-initio.

C. Loyola+. Comp. Mat. Sc, 49, 582 (2010).
Nanomaterials
They are materials with a grain size less than or equal to 100 nanometers (1 nm= 10E-9m). Massive interest in these materials arose due to certain physical properties, that demonstrated marked improvements over policrystalline materials or simple crystals of the same chemical composition, but conventional grain size (>μm). For example, nanostructured metals can resist greater stress than conventional materials, with thicker grain. Similarly, ceramics, usually brittle, show high hardness, high resistence to fractures and a superplastic behaviour when the grain size is refined to the nanometric regime. These materials are highly desired for applications that require extreme conditions of operation, such as those conditions experienced in the aeronautic industry, among others. There is a real need to investigate these materials at atomic level, both from experimental and theoretical perspectives.
Carbon Nanotubes
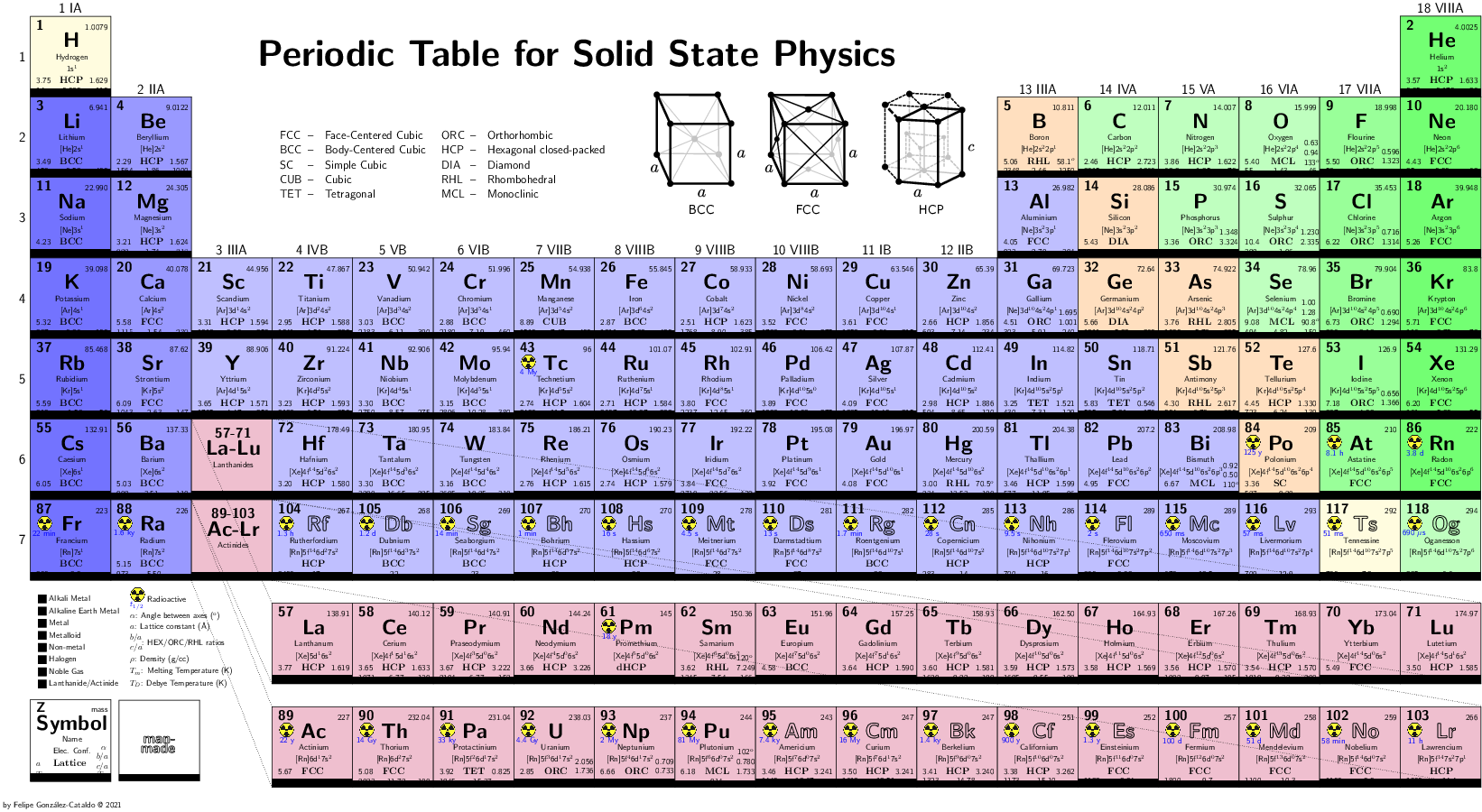
The carbon nanotubes are tubular structures composed of carbon atoms whose diameters are a few nanometers long, and lengths of micrometers long.
These structures are very studied nowadays, both theoretical and experimentally. due to their extraordinary structural and electronic properties that make them good candidates for a new generation of nanometric devices.
Some experiments have shown possible applications of these nanotubes in many systems, such as chemical and biological sensors, molecular wrappers, transistors, nanowires, and many others. In our group, there are two lines of theoretical research by means of computational quantum simulations on carbo nanotubes, these are:
- Electronic and structural properties of carbon nanotubes adsorbed in semiconductors surfaces, in particular, Si(001). We study the applications of the nanotubes to the current technology of silicon and its possible usage as an electronic device.
- Functionalization of the carbon nanotubes as chemical and biological sensors, as specific protein fixers for chemical bonds, and as ionic channels blockers. We investigate applications of nanotubes in biological systems.
Nano-Magnetism
We study the magnetic properties of materials at atomic level, via classical and ab-initio methods. With classical models we are studying heat transfer in unidimensional spin systems. According to our studies, these systems present certain anomalies in relation with the classical Fourier Law for systems of higher dimensions, like balistic, superdifusive or subdifusive behaviour.
Nano-Magnetism of First Principles
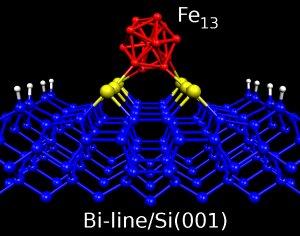
Self-organized structures over metalic and semiconductor surfaces such as islands, terraces or atomic lines have recieved a great deal of attention due to their possible applications as a pattern for magnetic growth of nanometric magnetic systems. In this field, in our group we investigate the adsorption of magnetic atoms and clusters in self-organized systems of low dimensionality formed over semiconductor surfaces.
In particular, we study lines of bismuth dimers that form over silicon surface Si(001). The mean width of these lines is 3nm, but their length can reach 500 nm long. Recently we have investigated the adsorption of Fe atoms over these Bi linesin Si(001). We are also studying the adsorption of little clusters of magnetic atoms over these lines and other sef-organized structures over surfaces, such as terraces.
Semiconductors Nano-Crystals
A semiconductor nanocrystal or quantum dot is a crystaline nanoscale structure, which is considered to have greater flexibility than other fluorescent materials, which make them accurate for nanoscale devices with computational applications where light is used to process information. The quantum attribute is used to remember that the behavior of the electron at such scales must be described in terms of quantum theory. Cadmium-Selenium nanostructures are an example of this type of crystal. The presence of a single electron in such structures can be used to store information.

Computational Simulations
By many scientists, considered as a "third methodology" of scientific research (in relation with theoretical and experimental approches). This method, complementary but many times alternative to the conventional ways of doing science, has exerted a big impact in almost all fields of science. The objective of the computational simulation is to solve the theoretical models in its overall complexity, by means of solving numerically the involved equations, making intensive and extensive use of the computers. The most common computational simulation methods used to study the physical properties of materials are Molecular Dynamics, Monte Carlo Method and Ab-Initio calculations.
Molecular Dynamics

This is a type of computational simulation that considers a system of atoms or molecules which are treated as a set of classic particles that interact by means of a interatomic potential. Due to the fact that molecular systems have a big number of particles, it is impossible to find properties of such complex systems analitically. Molecular Dynamics simulations allow us to solve this problem, using numerical methods. To obtain the trajectories in the phase space of the system (positions and velocities for all instants of time), Newton's equations of motions are solved numerically. Inside this scheme, many properties can be calculated, such as elastic properties, heat capacity, vibrational modes, diffusion constants, correlation functions and many others, that allow a direct comparison with experiments.
Ab-Initio Calculations

This method considers that a molecule or a solid is composed by ions and electrons, grouped in such a way that they form bonds between them. The physical properties that characterize the system depend on the different interactions between its components. These interactions are electronic in nature, and are described by the ion-ion and electron-electron interaction. The mechanics that goberns these systems at atomic level is quantum mechanics, and the equation to solve is the Schrödinger equation.
Our group studies the physical properties of solids by means of computational simulation using the molecular dynamics (MD) methods and ab-initio calculations. In DM computational programs have been used, such as Moldy (Molecular Dynamics) and GULP (General Utility Lattice Program). Nowadays, a more complete molecular dynamics code is being developed, a code that covers the necessities of our group. For ab-initio calculations, VASP, Quantum ESPRESSO and SIESTA programs have been used.
MAX Phases
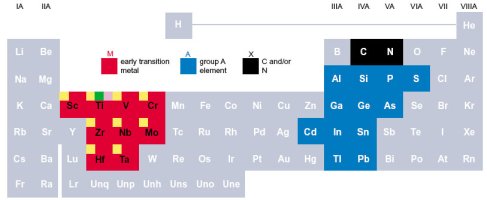
Max Phases materials are a type of ternary compounds in the form M_n+1 A X_n (MAX), where M is an early transition metal with N=1, 2 or 3. A is an element of the A group (mainly IIIA and IVA), and X is carbon (C) or nitrogen (N). These ternary carbides and nitrides combine unusual characteristics of metals and ceramics. The have high toughness but, at the same time, high ductibility.
Current Projects
- Project to Support the Development of International Cooperation for Research of Excellence (see)International Cooperation for simulation of nanobio systems.
- Project Bicentennial Ring-Chile ACT24 (see). Computational simulation laboratory in nanobio systems.
- AFOSR-USA Project FA9550-06-1-0540 (see)
- Fondecyt Project No. 1050197, Walter Orellana Interaction of carbon nanotubes with molecules and solids. A theoretical study.
Previous projects
- Fondecyt Project No. 1030063, Gonzalo Gutiérrez
- Study of the properties of materials through computational simulations: From the atomic to the nanoscopic.
- Study of properties of Materials by Computer Simulation: from atomic to nanoscopic regime
- Fondecyt Project No. 1050293, Eduardo Menéndez
- Physics of Novel Solar Cell Materials