

Areas de Investigación
Nuestro grupo lleva a cabo investigaciones en el área de materiales, nanomateriales y nanoestructuras. Nos interesa estudiar las propiedades estructurales, dinámicas, electrónicas y magnéticas de cerámicos, nanocristales, nanotubos, clusters y estructuras magnéticas de bajas dimensiones. Para ello, utilizamos técnicas de simulación computacional, tales como la teoría funcional de la densidad (DFT) para cálculos de estructura electrónica, así como dinámica molecular clásica y ab-initio.

C. Loyola+. Comp. Mat. Sc, 49, 582 (2010).
Nanomateriales
Son materiales con tamaño de grano menor o igual a los 100 nanómetros (1 nm= 10E-9m). El masivo interés por estudiar estos materiales se generó debido a que ciertas propiedades físicas mejoraron increíblemente respecto a materiales policristalinos o cristales simples de la misma composición química, pero de tamaño de grano convencional (>μm). Por ejemplo, los metales nanoestructurados pueden resistir esfuerzos mucho mayores que los metales convencionales, de grano más grueso. De modo similar, los cerámicos, normalmente quebradizos, muestran una alta dureza, alta resistencia a las fracturas, y un comportamiento superplástico, cuando el tamaño de grano es refinado al régimen nanométrico. Estos materiales son altamente deseados para aplicaciones que requieren condiciones extremas de operación, por ejemplo, son utilizados en la industria aeronáutica, entre otras. De allí la necesidad de investigar estos materiales, a nivel atómico, tanto desde el punto de vista experimental como teórico.
Nanotubos de Carbono
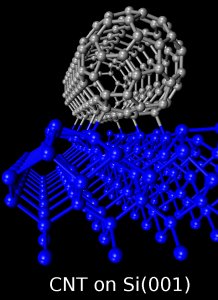
Son estructuras tubulares compuestas de átomos de carbonos cuyos diámetros son del orden de los nanómetros con longitudes que pueden llegar a los micrómetros.
Estas estructuras son actualmente materia de intensa investigación teórica y experimental debido a sus extraordinarias propiedades electrónicas y estructurales que los hacen promisores cadidatos para una nueva generación de dispositivos de escala nanométrica.
Experimentos han mostrado posibles aplicaciones de estos nanotubos en sistemas tan diversos como sensores químicos y biológicos, encapsuladores moleculares, transistores, nanohilos, entre otros. En nuestro grupo existen dos líneas de investigación teórica por medio de simulación computacional cuántica en nanotubos de carbono, estas son:
- Propiedades electrónicas y estructurales de nanotubos de carbono absorbidos en superficies semiconductoras, en particular Si(001). Aquí estudiamos la integración de los nanotubos de carbono a la actual tecnología del silicio y su posible uso como dispositivo electrónico.
- La funcionalización de nanotubos de carbono como sensores químicos y biológicos, como fijadores específicos de proteínas por enlaces químicos, y como bloqueadores de canales iónicos. Aquí investigamos el uso de nanotubos de carbono para aplicaciones en sistemas biológicos.
Nano-Magnetismo
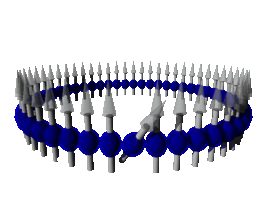
Estudiamos las propiedades magnéticas de materiales a nivel atómico tanto por métodos de cálculo ab-initio así como mediante cálculos modelos, ya sean cuánticos o clásicos. Respecto a los modelos clásicos, estamos investigando la trasmisión de calor en sistemas de espines unidimensionales. Según nuestros cálculos, estos sistemas presentan ciertas anomalías con respecto al modelo clásico de la Ley de Fourier para sistemas de dimensiones mayores, presentando comportamientos que pueden ser de tipo balístico, superdifusivo o subdifusivo.
Nano-Magnetismo de Primeros Principios
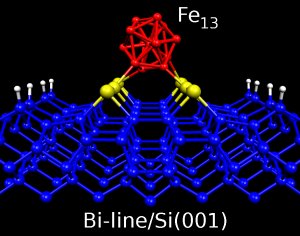
Estructuras auto-organizadas sobre superficies metálicas y semiconductoras tales como islas, terrazas o líneas atómicas han suscitado un gran interés debido a sus posibles aplicaciones como patrón para el crecimiento de sistemas magnéticos de escala nanométrica. Siguiendo esta línea, en nuestro grupo investigamos la absorción de átomos y clusters magnéticos sobre sistemas auto-organizados de baja dimensionalidad formados sobre superficies semiconductoras.
En particular, líneas de dímeros de bismuto que se forman sobre la superficie de silicio Si(001). Estas líneas tienen un ancho aproximado de 3 nm, con longitudes que pueden llegar a los 500 nm. Recientemente hemos investigado la absorción de átomos de Fe sobre estas líneas de Bi en Si(001). También estamos estudiando la absorción de pequeños aglomerados de átomos magnéticos (clusters) sobre estas líneas y otras estructuras auto-organizadas sobre superficies, tales como terrazas.
Nano-Cristales Semiconductores
Un nanocristal semiconductor o un punto cuántico es una estructura cristalina a nanoescala, la cual se considera que tiene una mayor flexibilidad que otros materiales fluorescentes, lo que lo hace apropiado para utilizarlo en construcciones a nanoescala de aplicaciones computacionales donde la luz es utilizada para procesar la información. El atributo cuántico sirve para recordar que el comportamiento del electrón en tales estructuras debe ser descrito en términos de la teoría cuántica. Las nanoestructuras de cadmio selenio son un ejemplo de este tipo de nanocristal. La presencia de un único electrón en tales estructuras puede ser utilizada para almacenar información.

Simulaciones Computacionales
Por muchos científicos es considerada “una tercera metodología” de la investigación científica. Este método, de carácter complementario y muchas veces alternativo a los modos convencionales de hacer ciencia, el experimental y el teórico, ha ejercido un fuerte impacto en prácticamente todos los campos de la ciencia. El objetivo de la simulación computacional es resolver los modelos teóricos en su total complejidad, mediante la resolución numérica de las ecuaciones involucradas, haciendo uso intensivo (y extensivo) de computadores. Los métodos de simulación computacional comúnmente usados para el estudio de las propiedades físicas de los materiales, son los de Dinámica Molecular, Método Montecarlo y Cálculos ab-initio.
Dinámica Molecular

Es un tipo de simulación computacional que considera un sistema de átomos o moléculas los cuales son tratados como un conjunto de partículas clásicas que interactúan unas con otras mediante un potencial interatómico. Debido a que en general los sistemas moleculares poseen un gran número de partículas, es imposible encontrar propiedades de tales sistemas complejos analíticamente. La simulación de Dinámica Molecular resuelve este problema usando métodos numéricos. Para obtener las trayectorias del espacio de fase del sistema (posiciones y velocidades de todos los átomos en todos los tiempos) se resuelven las ecuaciones de Newton numéricamente. Dentro de este esquema se pueden calcular propiedades elásticas, capacidad calórica, modos vibracionales, constantes de difusión, funciones de correlación y tantas otras que permiten una comparación directa con los experimentos.
Calculos Ab-Initio
Este método considera que una molécula, o un sólido está compuesto por iones y electrones, agrupados de tal modo que forman enlaces entre ellos. Las propiedades físicas que caracterizan al sistema dependen de las diferentes interacciones entre sus componentes. Estas interacciones son de carácter eléctrico, y quedan descritas por la interacción ion-ion, ion-electrón y electrón-electrón. La mecánica que gobierna al sistema a nivel atómico es la mecánica cuántica, y lo que debe resolverse es la ecuación de Schrödinger.
Nuestro grupo estudia las propiedades físicas de los sólidos mediante simulación computacional a través de los métodos de dinámica molecular (DM) y cálculos ab-initio. En DM se han utilizado los programas computacionales Moldy (Molecular Dynamics) y GULP (General Utility Lattice Program). Actualmente se está desarrollando un código de dinámica molecular más completo, que abarque las necesidades de nuestro grupo. Para cálculos ab-initio se han utilizado los programas VASP, Quantum ESPRESSO y SIESTA.
MAX Phases
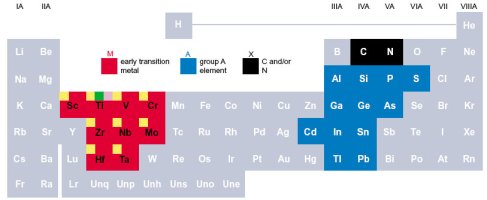
Los materiales max phases son una clase de compuestos ternarios de la forma: M_n+1 A X_n (MAX), donde M es un metal de transición temprana con N = 1, 2 o 3, A es un elemento del grupo A (sobre todo IIIA e IVA), y X es o C y/o N. Estos carburos y nitruros ternarios combinan características inusuales de los metales y de los cerámicos. Poseen alta dureza, pero al mismo tiempo gran ductibilidad.
Proyectos en Curso
- Proyecto de Apoyo al Desarrollo de la Cooperación Internacional para la Investigación de Excelencia (ver)Cooperación Internacional para simulación de nanobio sistemas.
- Proyecto Anillo Bicentenario-Chile ACT24 (ver). Laboratorio de simulación computacional in nanobio sytems.
- Proyecto AFOSR-USA FA9550-06-1-0540 (ver)
- Proyecto Fondecyt No. 1050197, Walter Orellana Interacción de nanotubos de carbono con moléculas y sólidos. Un estudio teórico.
Proyectos Anteriores
- Proyecto Fondecyt No. 1030063, Gonzalo Gutiérrez
- Estudio de las propiedades de materiales mediante simulaciones computacionales: De lo atómico a lo nanoscópico.
- Study of properties of Materials by Computer Simulation: from atomic to nanoscopic regime
- Proyecto Fondecyt No. 1050293, Eduardo Menéndez
- Physics of Novel Solar Cell Materials